Integrative Genomics Analyses using the gact and qgg R packages
Center for Quantitative Genetics and Genomics, Aarhus University, Denmark
Why Integrate Diverse Data Sources?
- Complex traits arise from interacting molecular layers — genetic, transcriptomic, proteomic, and metabolic.
- Single data types provide only partial insights complex biological systems (GWAS: DNA → disease phenotype)
- Integration connects variants → genes → pathways → phenotypes may help reveal molecular mechanisms that drive traits and diseases.
- Enables functional interpretation, better prediction, and new discoveries across molecular systems.
From Data Integration to Discovery
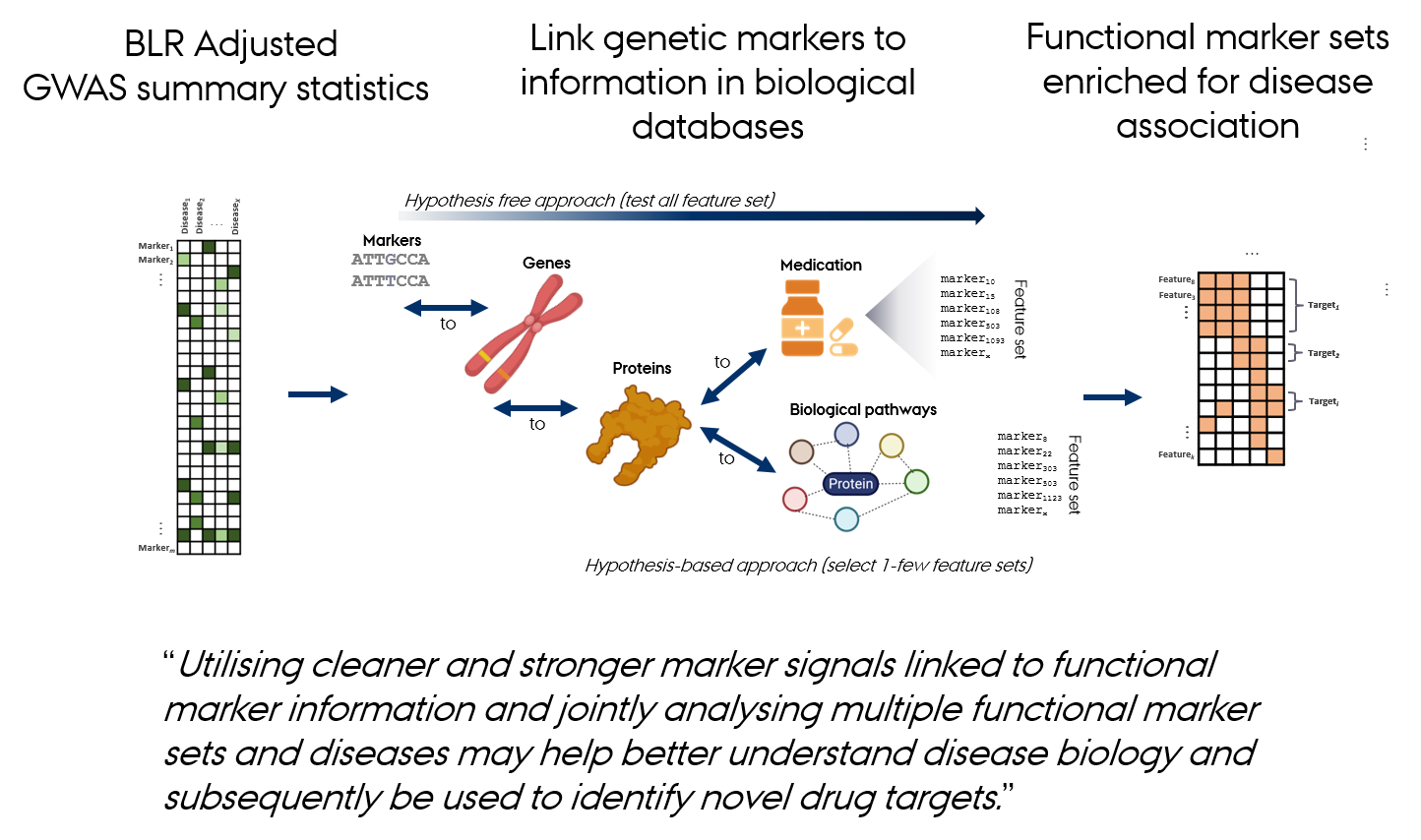
The gact R Package
gact provides an infrastructure for efficient processing of large-scale genomic association data, with core functions for:
- Establishing and populating a database of genomic associations
- Downloading and processing biological databases
- Handling and processing GWAS summary statistics
- Linking genetic markers to genes, proteins, metabolites, and biological pathways
- Integrates with statistical machine learning tools in the qgg R package
gact is intended to serve as a practical implementation of integrative genomics, bridging statistical modeling and biological interpretation, and supporting reproducible and extensible workflows.
Integrating Data with gact
The gact() function is a single R command that creates and populates the Genomic Association of Complex Traits (GACT) database.
It automates three main tasks:
- Infrastructure creation – sets up a structured folder-based database
(glist,gstat,gsets,marker,gtex,download, etc.) - Data acquisition – downloads and organizes multiple biological data sources
(e.g., GWAS Catalog, Ensembl, GTEx, Reactome, STRING, STITCH, DGIdb) - Marker and feature set generation - integrates data across sources to create curated genomic feature sets that form the basis for the integrative genomic analyses.
Biological Databases Used by gact
gact constructs gene and marker sets from a wide range of curated biological databases:
- Ensembl — genes, transcripts, and proteins
- Ensembl Regulation — regulatory genomic features
- GO, Reactome, KEGG — ontology and pathway sets
- STRING, STITCH — protein and chemical complexes
- DrugBank, ATC — drug–gene and drug-class associations
- DISEASE — disease–gene associations
- GTEx — eQTL-based gene sets
- GWAS Catalog — trait-associated variants and genes
- VEP — functional variant annotations
We plan to add additional biological resources in gact.
From Database to Model Inputs
The gact R package includes utility functions to extract and structure data from the GACT database into analysis-ready inputs — \(\mathbf{Y}\) (e.g., summary statistic outcomes) and \(\mathbf{X}\) (genomic or biological features).
getMarkerStat()— retrieve GWAS summary statistics (Y’s)
getFeatureStat()— extract gene-, protein-, or pathway-level results (Y’s)
getMarkerSets()— define biological groupings (basis for X’s)
designMatrix()— build feature matrices (X) linking variants or genes to biological feature sets
Together, these functions form a reproducible workflow for generating standardized input data for Bayesian Hierarchical Models and other machine learning approaches.
The qgg R Package
qgg provides tools for statistical modeling and analysis of large-scale genomic data, including:
- Fine-mapping of genomic regions using Bayesian Linear Regression (BLR) models
- Polygenic scoring using Bayesian Linear Regression (BLR) models
- Gene set enrichment analysis using Bayesian Linear Regression (BLR) models
qgg handles large-scale genomic data through efficient algorithms and sparse matrix techniques, combined with multi-core processing using OpenMP, multithreaded matrix operations via BLAS libraries (e.g., OpenBLAS, ATLAS, or MKL), and fast, memory-efficient batch processing of genotype data stored in
binary formats such as PLINK .bed files.
Tutorials using the qgg and gact R packages
The tutorials listed below are external resources and are not included in this repository.
Gene analysis using VEGAS: Gene analysis using the VEGAS (Versatile Gene-based Association Study) approach using the 1000G LD reference data processed above,
Gene set analysis using Bayesian MAGMA: Pathway prioritization using a single and multiple trait Bayesian MAGMA models and gene-level statistics derived from VEGAS (Gholipourshahraki et al.2024).
Gene ranking using PoPS: Polygenic Prioritization Scoring (PoPS) using BLR models and gene-level statistics derived from VEGAS (work in progress).
Finemapping using BLR models: Finemapping of gene and LD regions using single trait Bayesian Linear Regression models (Shrestha et al.2025).
Polygenic scoring using BLR models: Polygenic scoring (PGS) using Bayesian Linear Regression models and biological pathway information (work in progress).
Polygenic scoring using PGS Catalog: Polygenic scoring (PGS) using summary statistics from PGS catalog and biological pathway information.
LD score regression: LD score regression for estimating genomic heritability and correlations.
From Data Integration to Modeling
The gact and qgg R packages bridges data integration, statistical modeling and biological interpretation, enabling reproducible and extensible workflows.
- Integrates biological information across molecular layers — from genome to pathways, complexes, and drug–gene interactions
- Uses structured priors and hierarchical modeling to share information, regularize effect estimates, and quantify uncertainty
- Enables data-driven discovery and prediction
Next Steps
- Apply the framework to new and relevant use cases
- Expand integration with more biological data resources
- Incorporate additional statistical and machine learning methods
Further Reading and Collaboration
💡 We are open to collaboration!
If you’re interested in applying BLR methods or contributing to the gact framework, please reach out.
Further Reading