Introduction to Population Genetics
Chapter 18 - Introduction to Genetic Analysis (12th ed.)
2026-03-27
Visualizing Genetic Variation
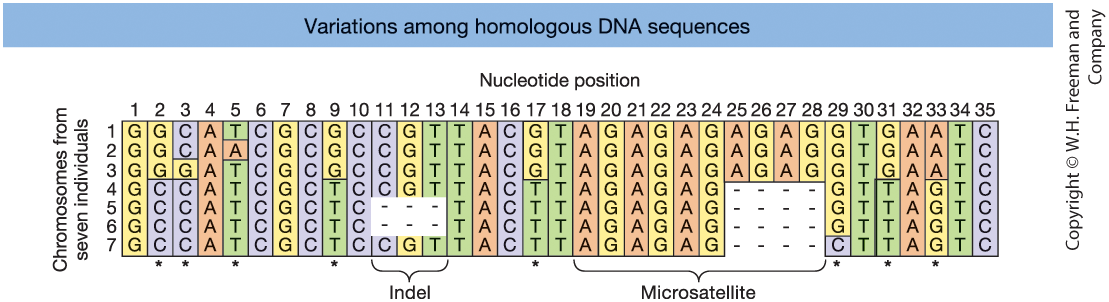
Figure 18-1. Aligned DNA sequences from seven chromosomes. Asterisks indicate SNPs. Indels and a microsatellite region are also shown.
From DNA to Genotype Data
Genomic technologies allow measurement of variation at thousands to millions of loci.
Marker-based genotyping
- Assay predefined variants
(SNP arrays, STR panels)
- Efficient for large sample sizes
Sequencing-based approaches
- Discover and genotype variants simultaneously
- Provide more comprehensive genomic information
Large genotype datasets across many loci form the foundation of population genetic analysis.
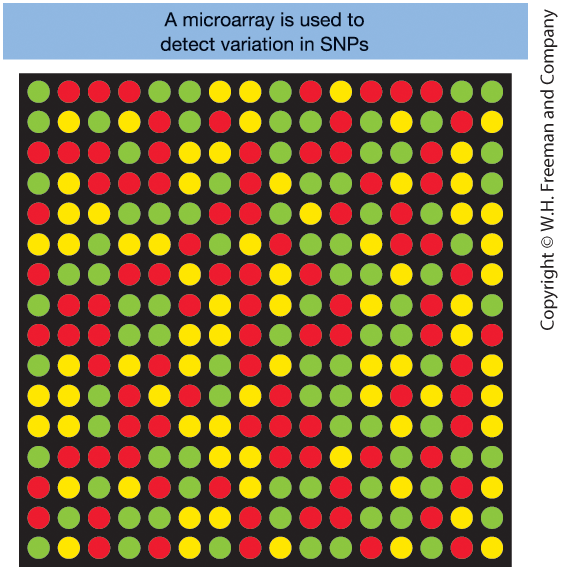
Figure 18-2. Microarray for SNP genotyping
- Each dot = one SNP
- Red/green = homozygous
- Yellow = heterozygous
Visualizing Haplotypes and Their Relationships
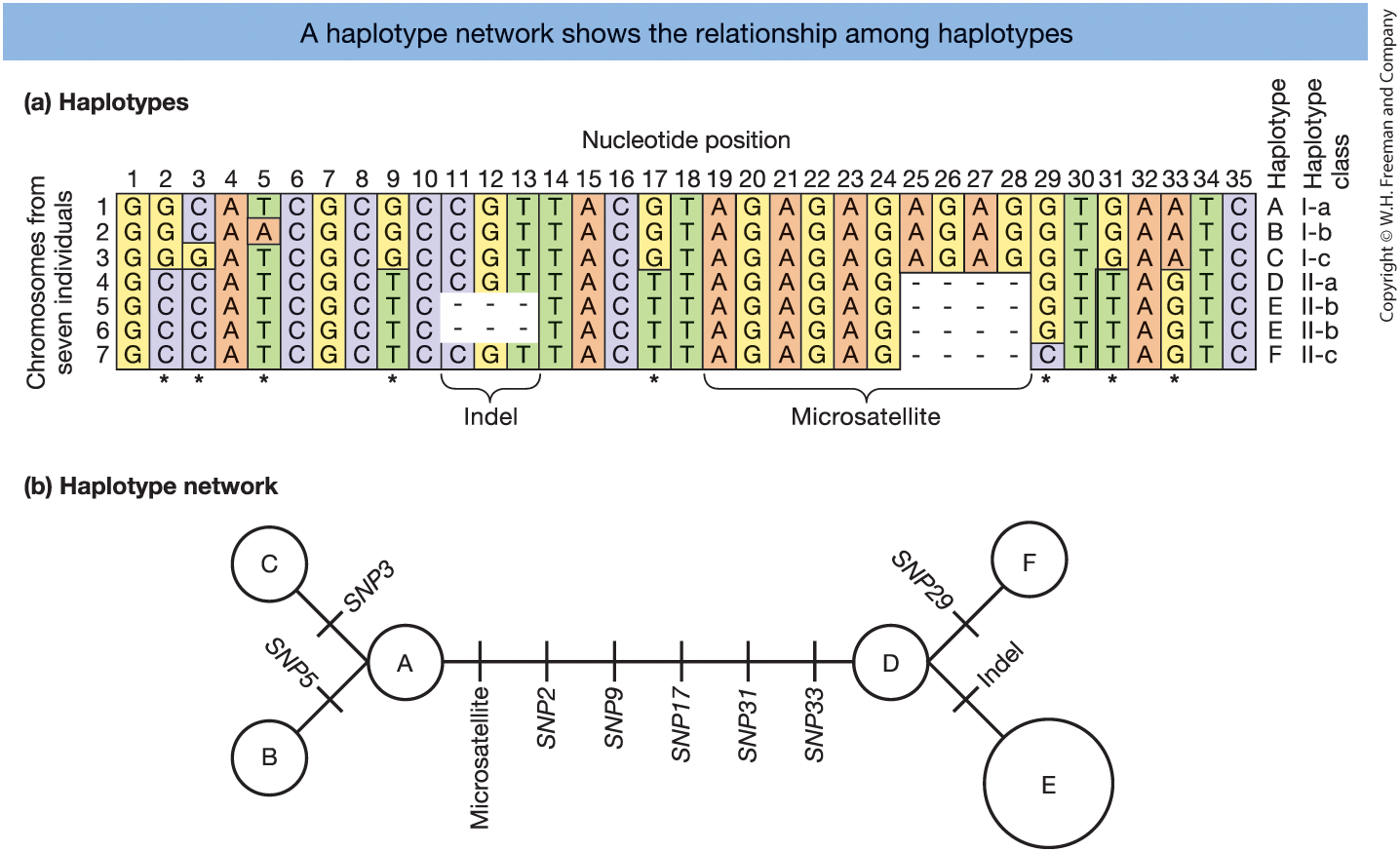
Figure 18-4.
(a) Six haplotypes (A–F) from aligned DNA sequences.
(b) Haplotype network showing mutational relationships.
- Circles = haplotypes
- Branches = mutations
Haplotype Network for Human mtDNA
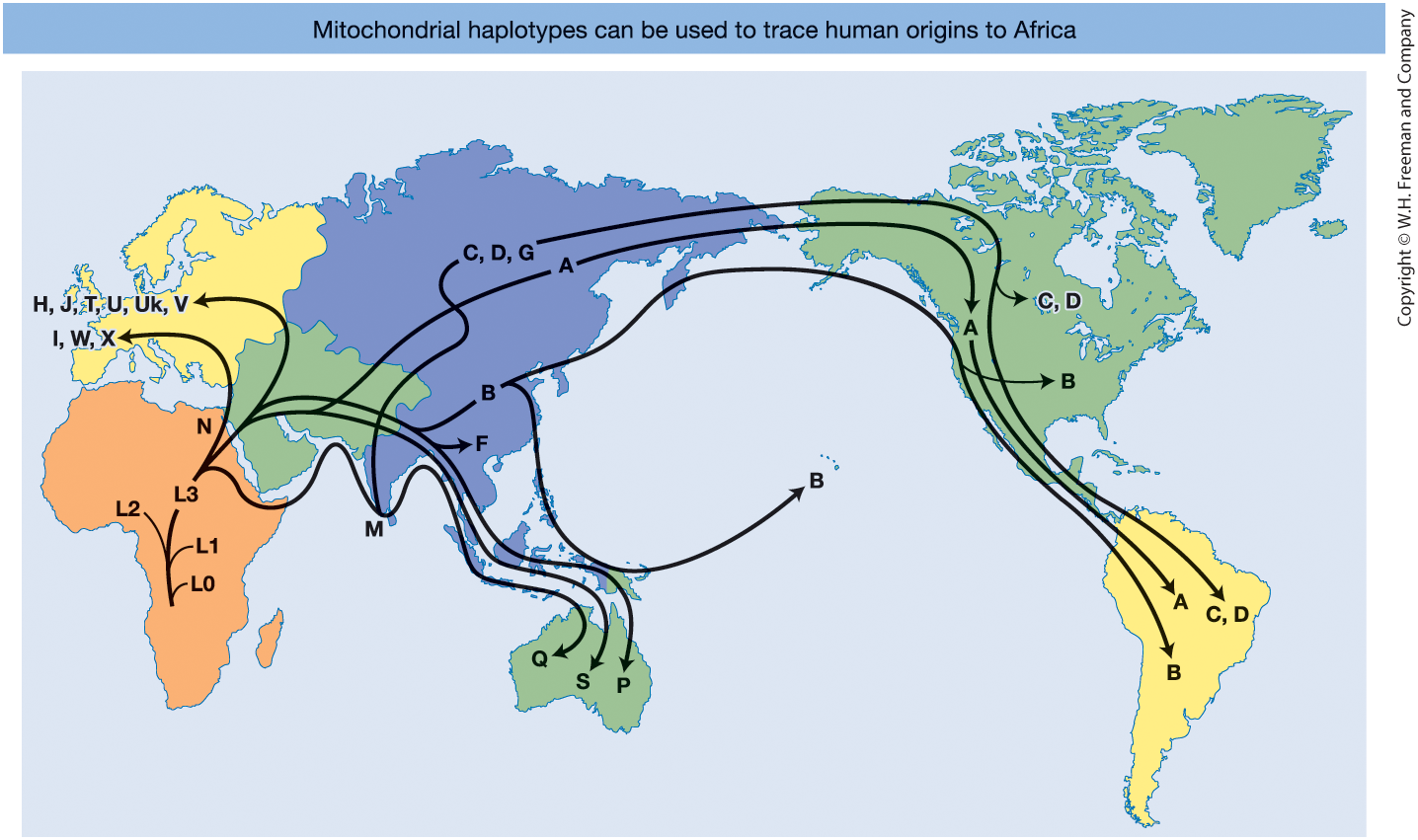
Figure 18-6 Human mitochondrial DNA haplotype network mapped globally.
- The ancestral L haplogroup appears in Africa
- Derived haplogroups spread worldwide
- Supports the Out of Africa model
The Gene Pool Concept
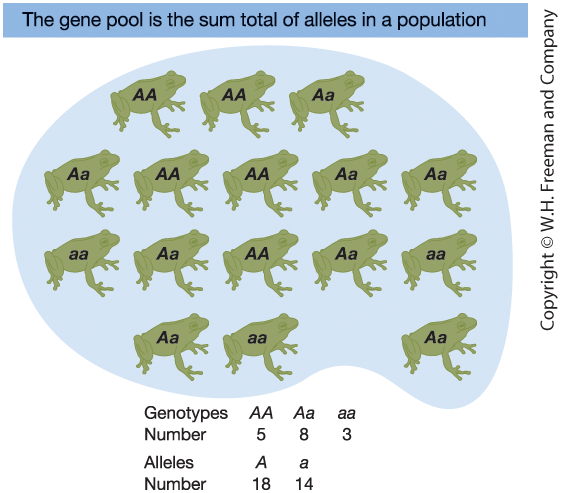
Figure 18-7. A Frog Gene Pool
Gene pool = total collection of alleles
in a population at a given time.
Population size: N = 16 (diploid)
Total alleles at one locus: 2N = 32
Genotype counts:
- 5 AA
- 8 Aa
- 3 aa
Allele counts:
- 18 A
- 14 a
Geographic Gradients in Allele Frequency
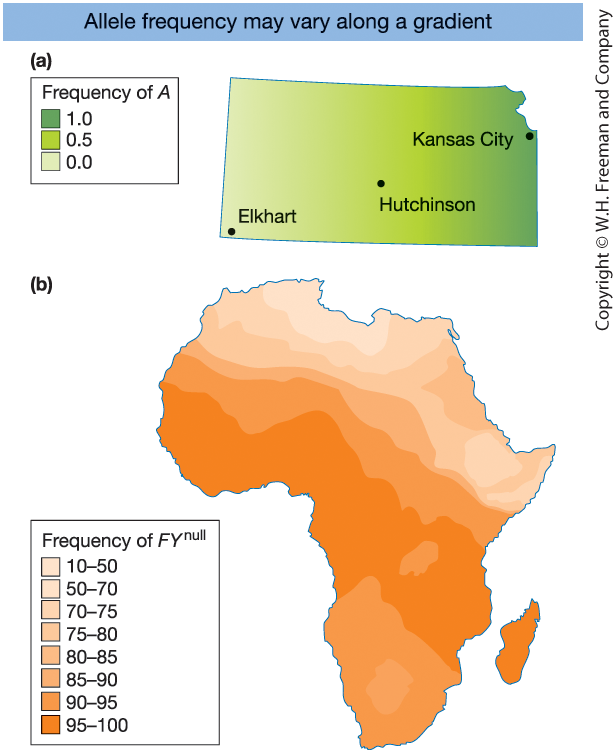
Figure 18-11
- Hypothetical sunflower allele frequency gradient (Kansas)
- Duffy blood group allele (\(FY^{0}\)) across Africa
\(\Rightarrow\) Geographic variation creates
population structure
Local HWE ≠ global HWE
Inbreeding and Identity by Descent (IBD)
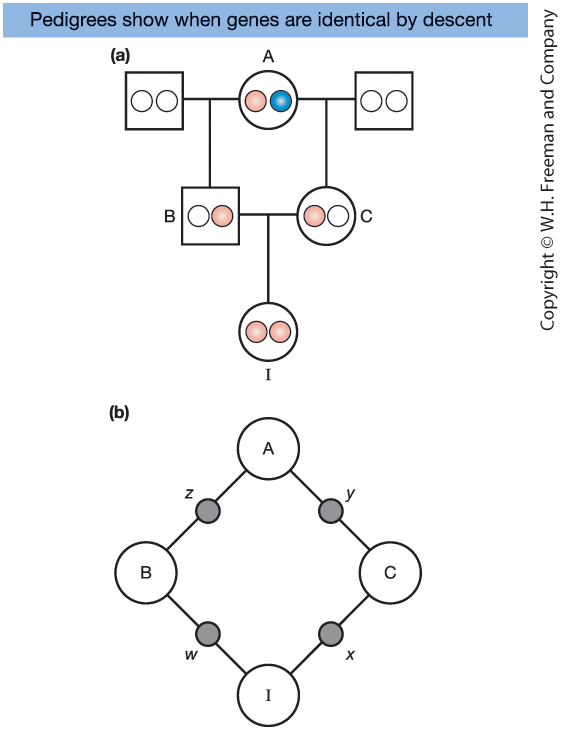
Consider the half-sib pedigree in Figure 18-12:
- B and C are half-siblings
- They share a common mother, A
- Their daughter is I
A has two gene copies:
- One from her mother (pink)
- One from her father (blue)
I is inbred because there is a closed ancestral loop.
If I’s two alleles trace back to the same physical gene copy in A, they are identical by descent (IBD).
The probability of this event is the inbreeding coefficient, \(F_I\).
Inbreeding and Genetic Disorder Risk
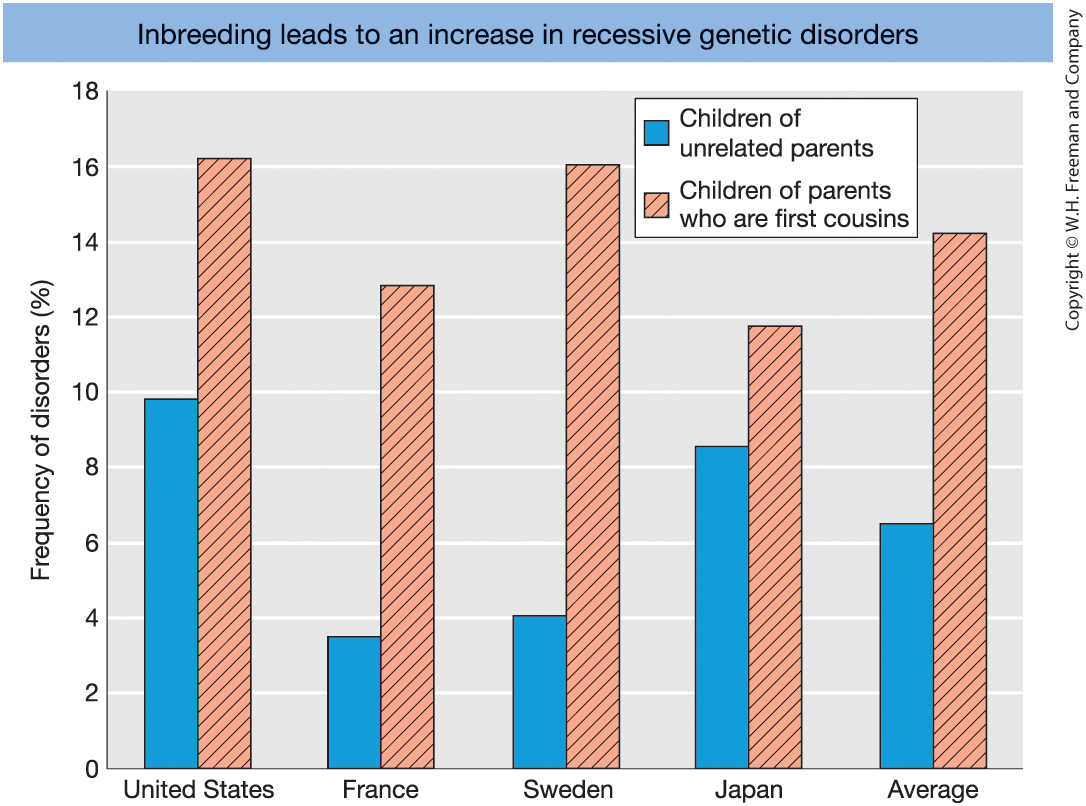
Figure 18-13 Frequency of genetic disorders among children of unrelated parents (blue columns) compared to that of children of parents who are first cousins (red columns with diagonal lines). [Data from C. Stern, Principles of Human Genetics, W. H. Freeman, 1973.]
Inbreeding Increases in Small Populations
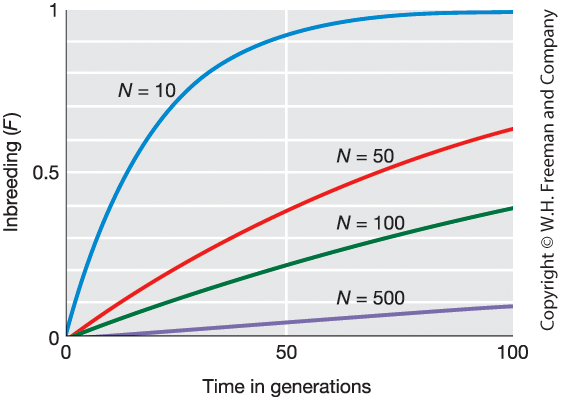
Figure 18-13
Increase in inbreeding coefficient (\(F\)) over generations for different population sizes (\(N\)).
Smaller \(N\) \(\Rightarrow\) Faster increase in \(F\)
Nucleotide Diversity Across Species
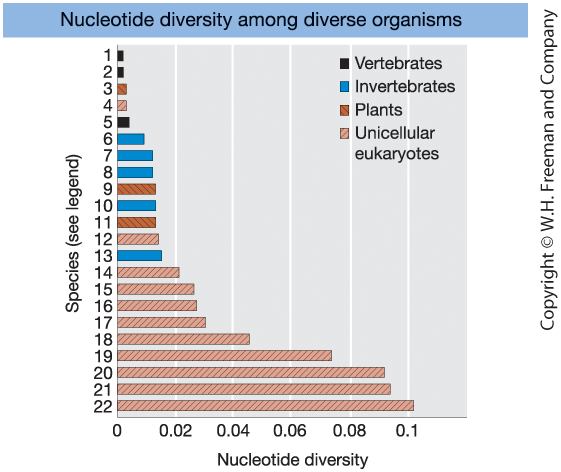
Figure 18-15
Levels of nucleotide diversity
at synonymous (silent) sites
in diverse organisms.
Key pattern:
- Vertebrates \(\Rightarrow\) low diversity
- Invertebrates & plants \(\Rightarrow\) moderate
- Many unicellular eukaryotes \(\Rightarrow\) high
Migration and Genetic Admixture
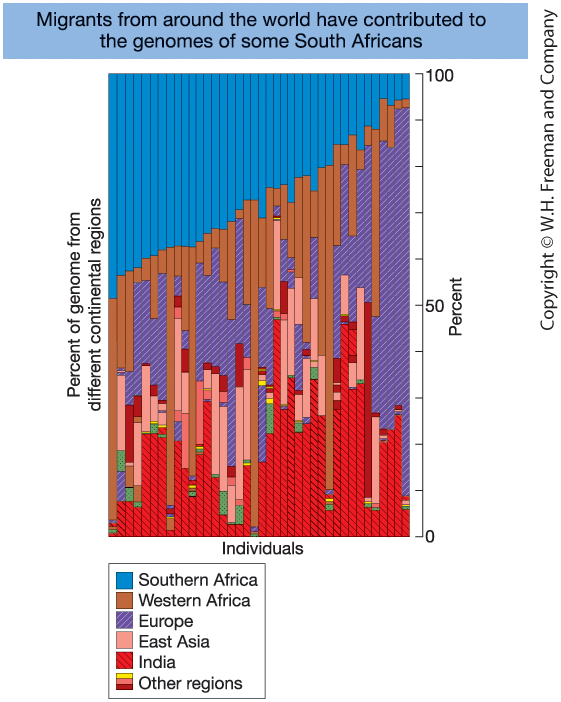
Figure 18-16. Genetic admixture in individuals of mixed ancestry (South Africa).
Each vertical bar represents one individual.
Colors indicate genomic segments inherited
from different ancestral populations.
Admixture reflects historical migration
and interbreeding between populations.
Random Genetic Drift: Simulation Results
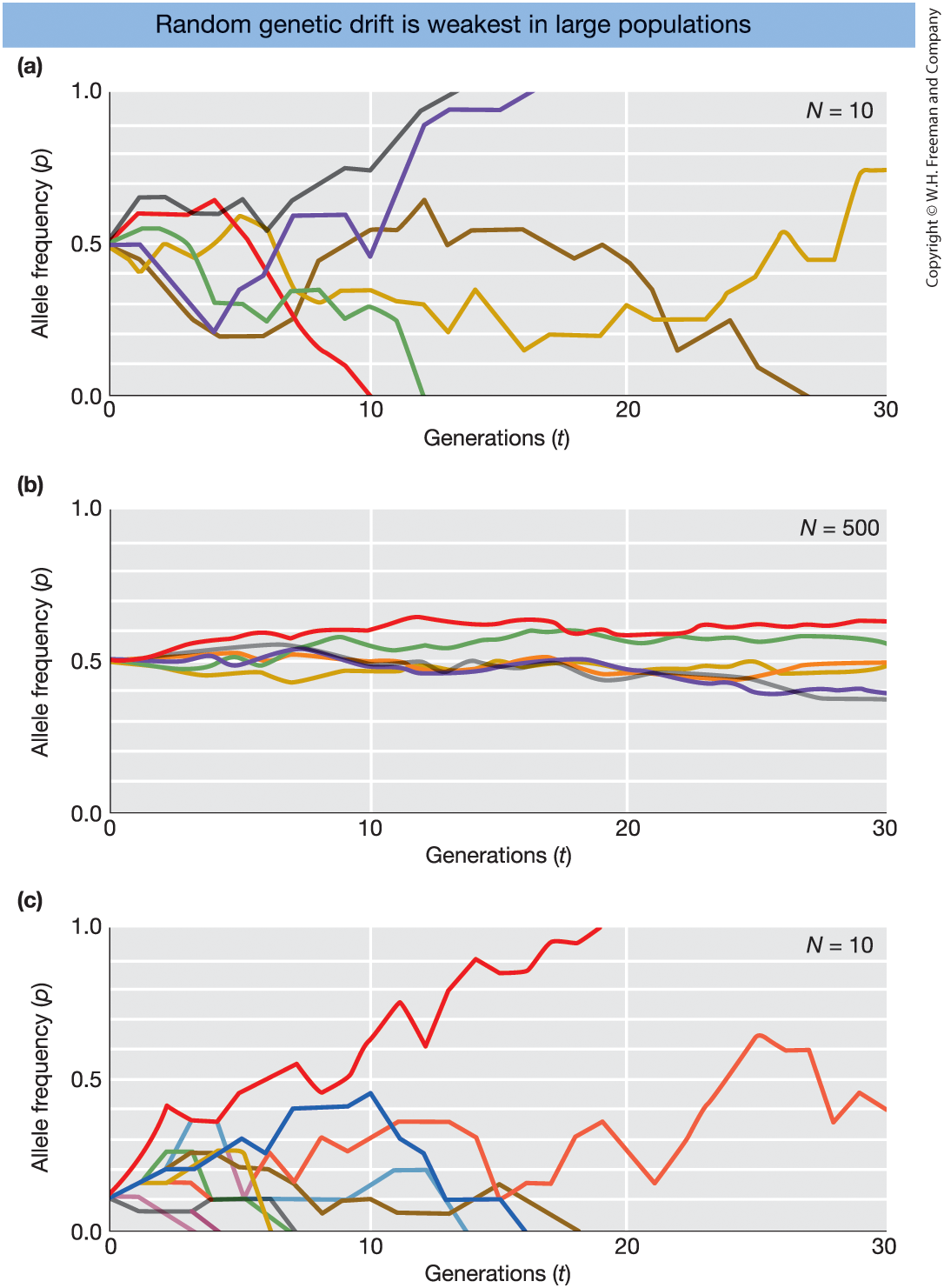
Figure 18-18
Simulations of random genetic drift
Each colored line = one simulated population
tracked for 30 generations.
- Small population (\(N = 10\), \(p_0 = 0.5\))
- Large population (\(N = 500\), \(p_0 = 0.5\))
- Small population (\(N = 10\), \(p_0 = 0.1\))
Hardy–Weinberg assumes an infinitely large population.
Real populations are finite.
In finite populations, allele frequencies change by chance:
Random genetic drift
Selection and Allele Frequency
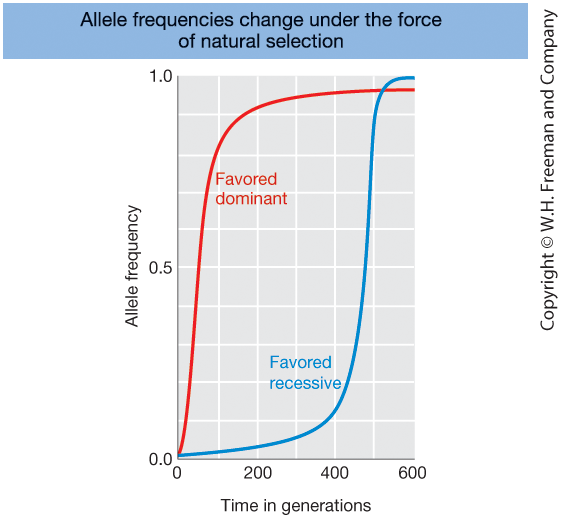
Figure 18-21
Change in allele frequency over time for:
- Favored dominant allele (red)
- Favored recessive allele (blue)
Both alleles increase due to natural selection, but at different rates.
Selective Sweep and Genetic Variation

Figure 18-22
Haplotypes before and after
a beneficial allele (red)
sweeps to fixation.
After selection:
- Reduced genetic diversity
- Increased linkage disequilibrium
- One dominant haplotype
Selective Sweep and Genetic Variation
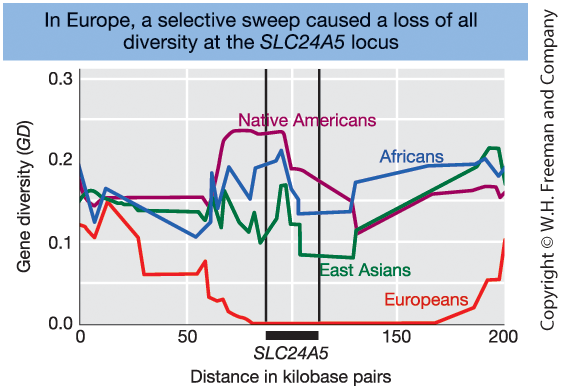
Figure 18-23. Gene diversity near the SLC24A5 locus on chromosome 15.
Gene diversity is strongly reduced in Europeans
around the SLC24A5 locus.
This pattern is consistent with
a recent selective sweep.
Balancing Selection Maintains Genetic Diversity
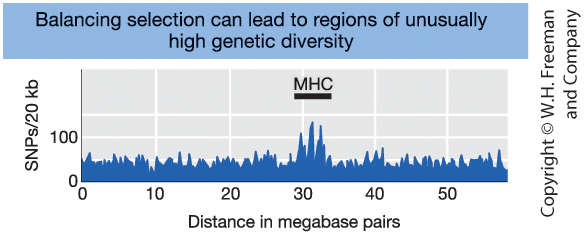
Figure 18-24
Number of SNPs along chromosome 6.
The MHC region shows a pronounced spike
of unusually high genetic diversity.
Key idea:
Balancing selection can maintain multiple alleles at a locus.