Introduction to Quantitative Genetics
2026-03-27
Natural and Artificial Selection
Natural selection and selective breeding can both cause changes in animals and plants:
Natural selection happens naturally.
Selective breeding occurs when humans intervene => artificial selection.
Selective breeding (and natural selection) takes place over many generations.

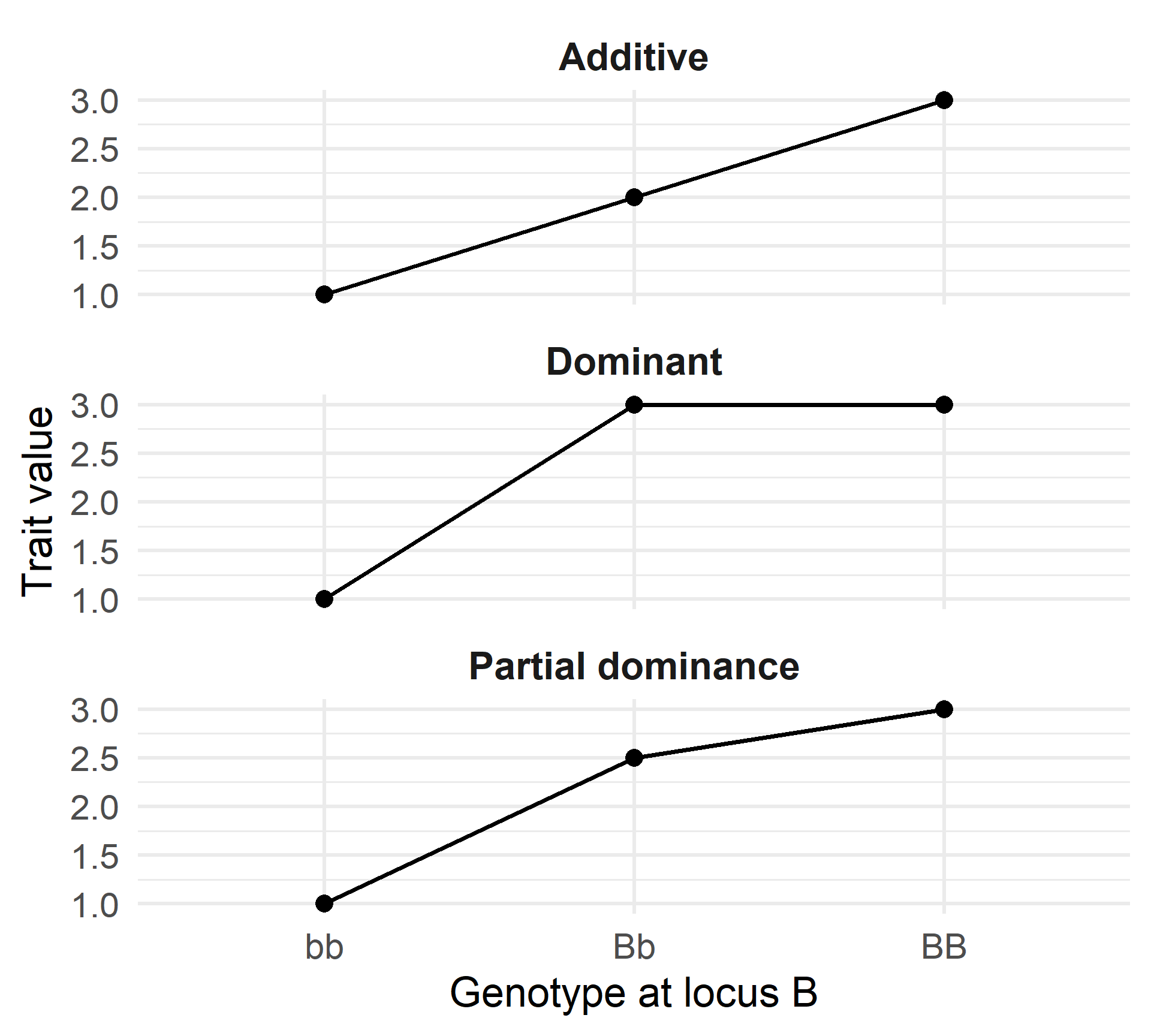
What Are Complex (Quantitative) Traits?
Mendelian traits
- Discrete phenotypes (e.g., red/white, sick/healthy)
- Simple genotype–phenotype relationship
- Often large gene effects
Quantitative traits
- Many phenotype classes, often continuously distributed
- Influenced by many genes, each with small effects
- Individual gene effects are typically not directly observable
- Often influenced by the environment
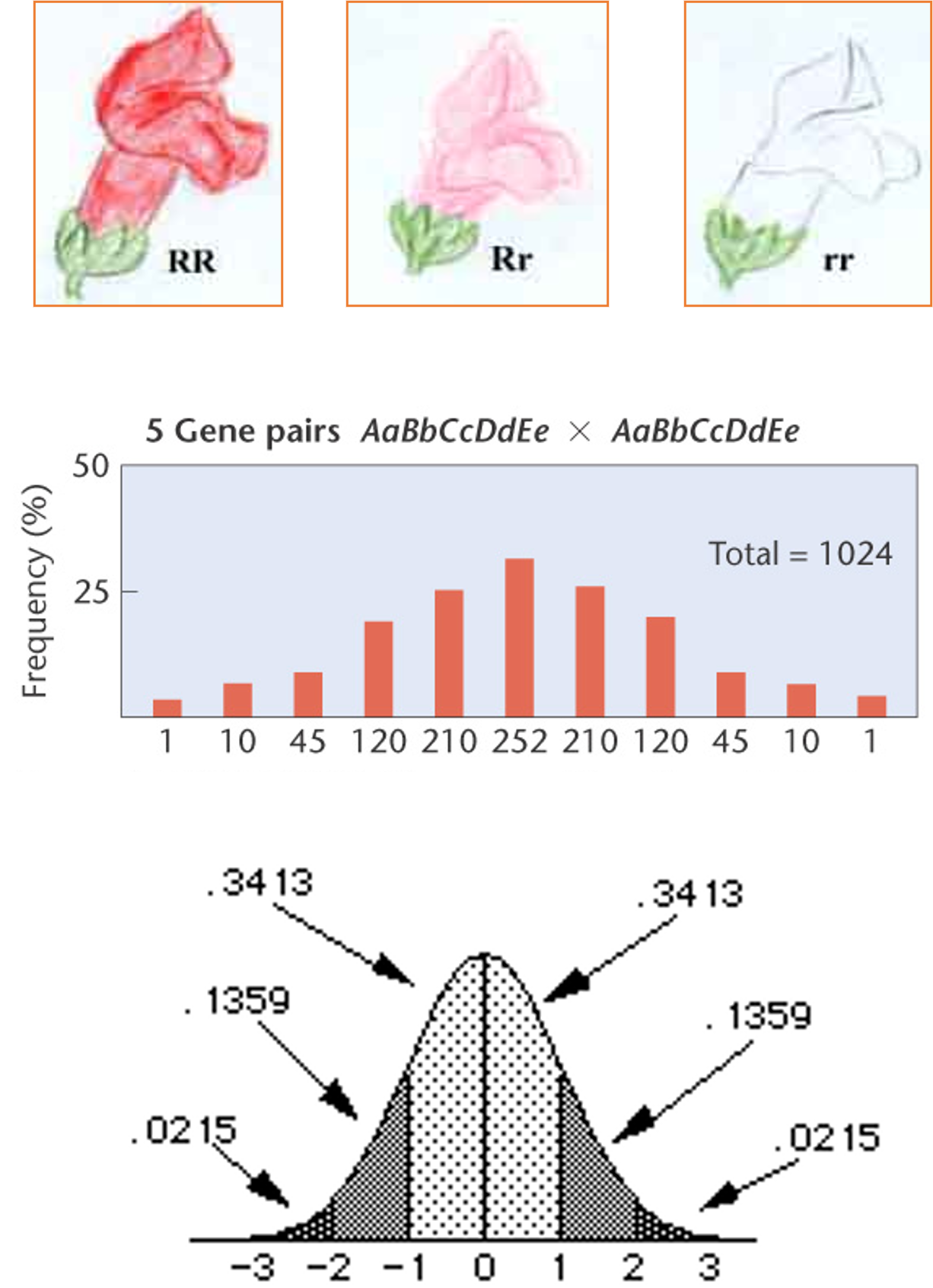
Normal Distribution (Effect of μ and σ)
Genotype by Genotype Interaction (Epistasis)
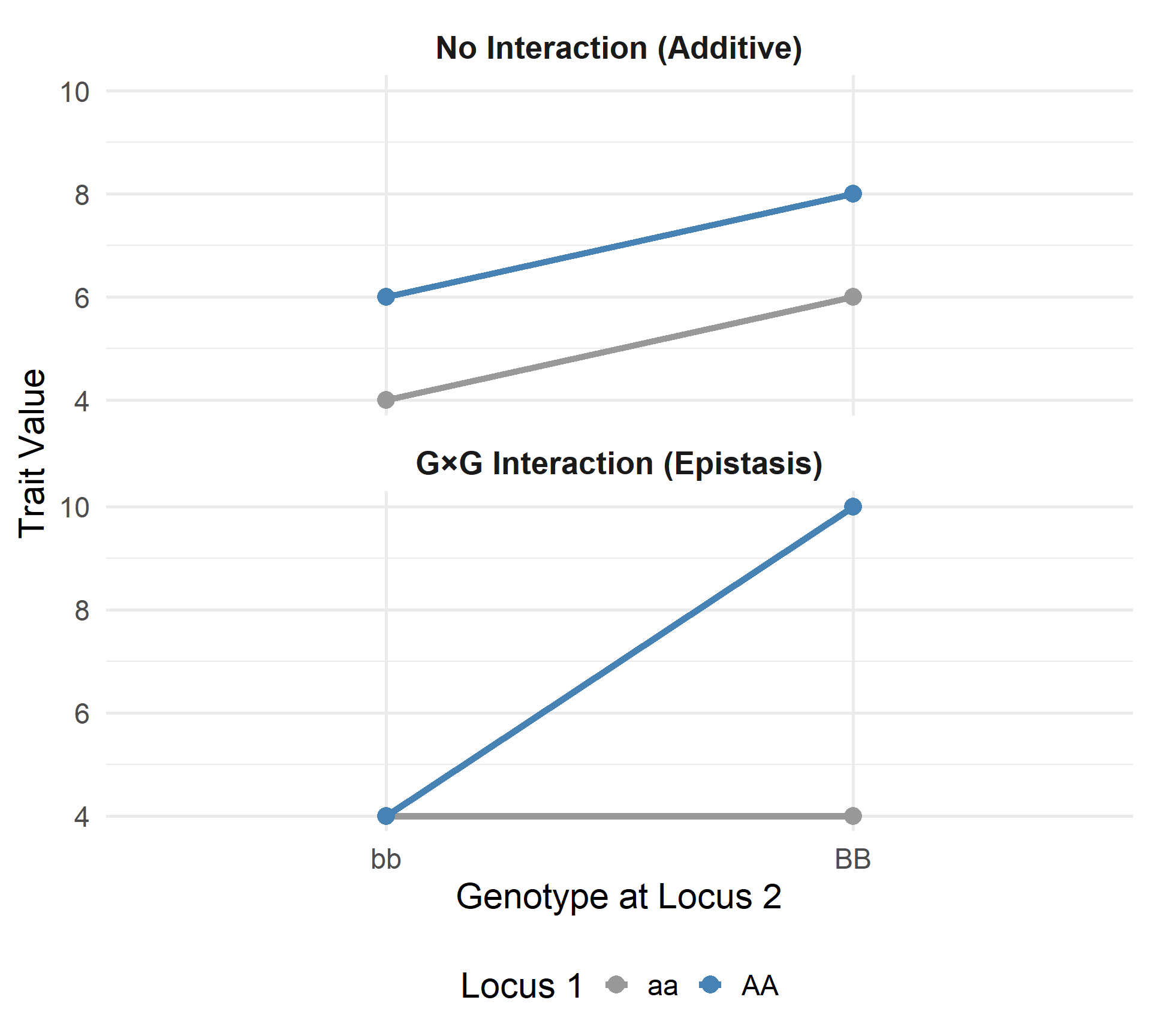
Interaction between Locus 1 and Locus 2
No Interaction (Additive)
- Parallel lines: The lines do not cross or diverge.
- The effect of \(AA\) is the same regardless of whether the background is \(bb\) or \(BB\).
- Genetic effects at different loci simply add up linearly.
G\(\times\)G Interaction (Epistasis)
- Non-parallel lines: Lines cross or show different slopes.
- The effect of \(AA\) depends on the genotype at Locus 2.
- Example: \(AA\) only increases the trait value if the individual also carries \(BB\).
Key Idea:
The phenotypic expression of one gene is modified by the presence of another gene.
Genotype × Environment Interaction
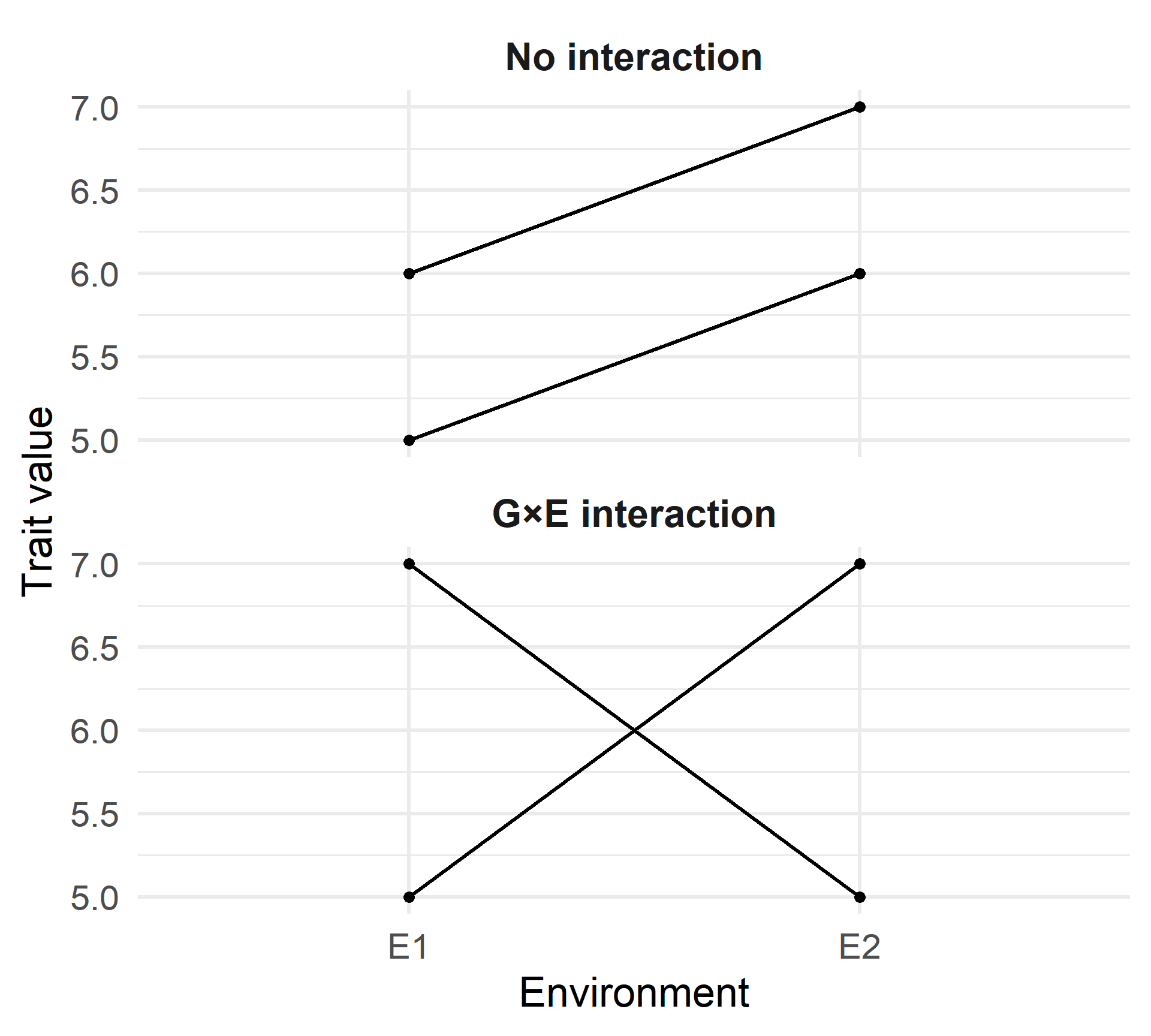
No interaction
- Parallel lines
- Genotype difference is constant across environments
G×E interaction
- Lines cross (or diverge)
- Genotype ranking depends on environment
Key idea:
Genotype performance can be environment-dependent.
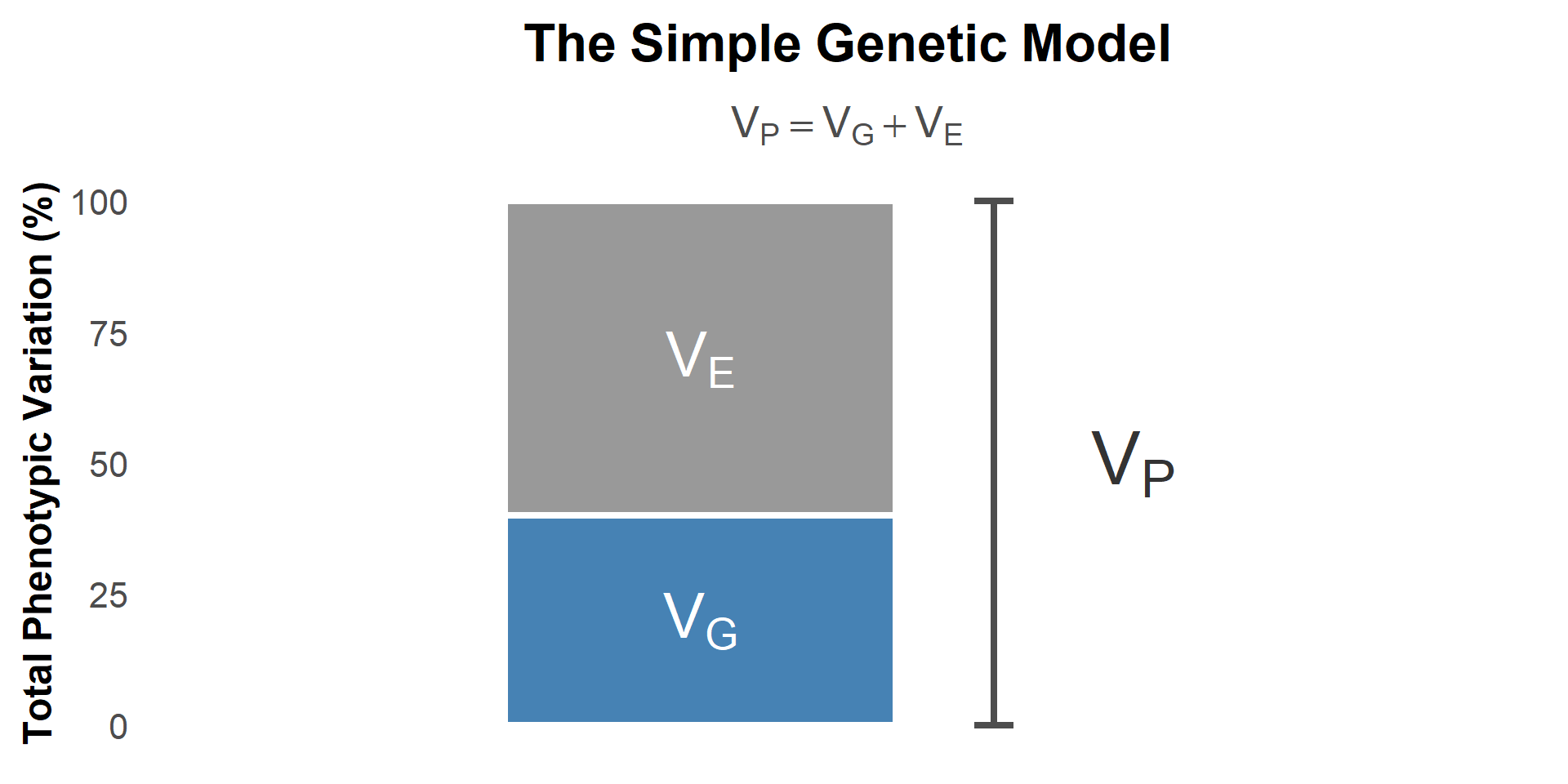
Visualizing Variance Partitioning
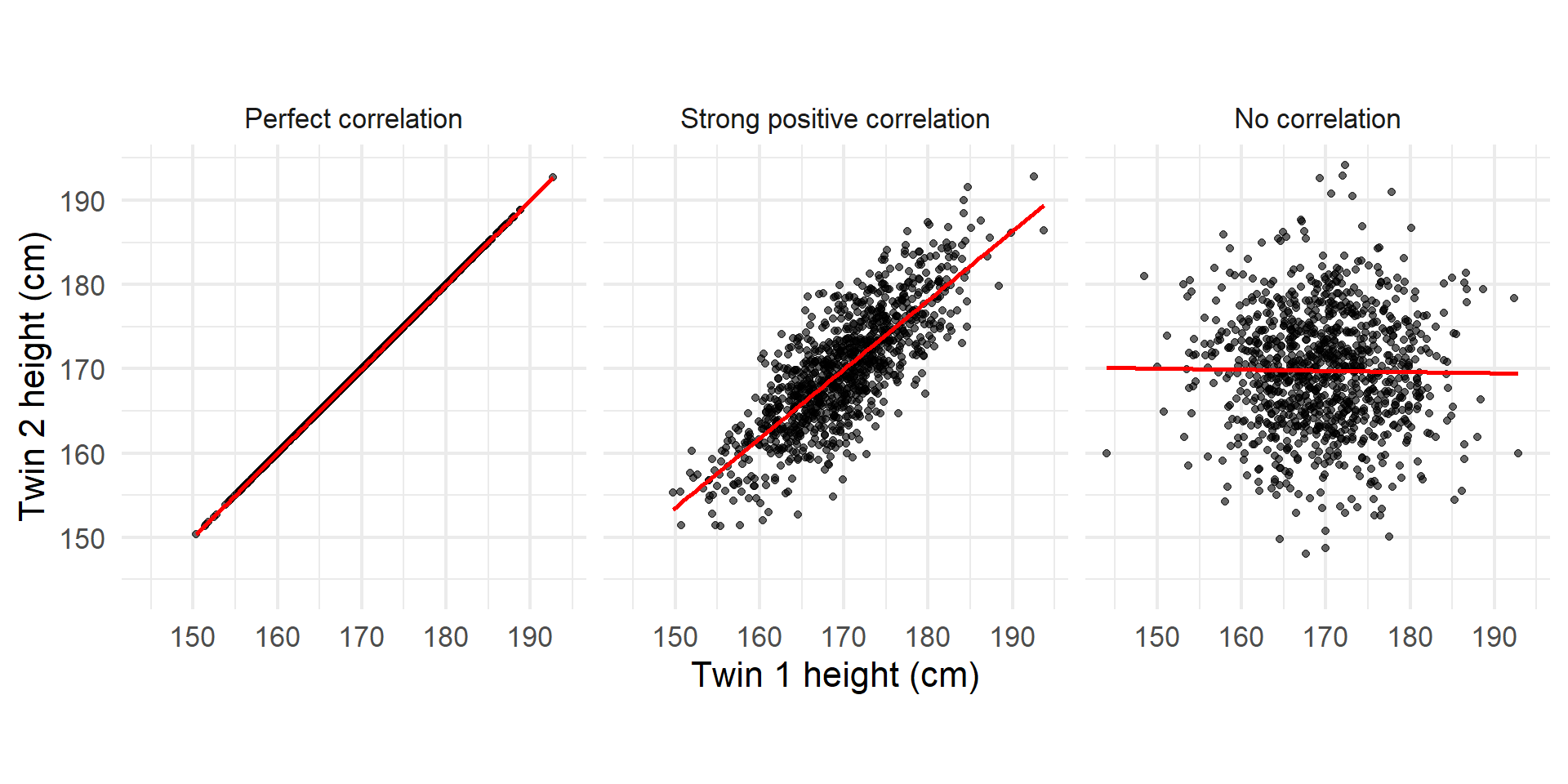
Visualizing Correlation
Narrow-Sense Heritability (h²)
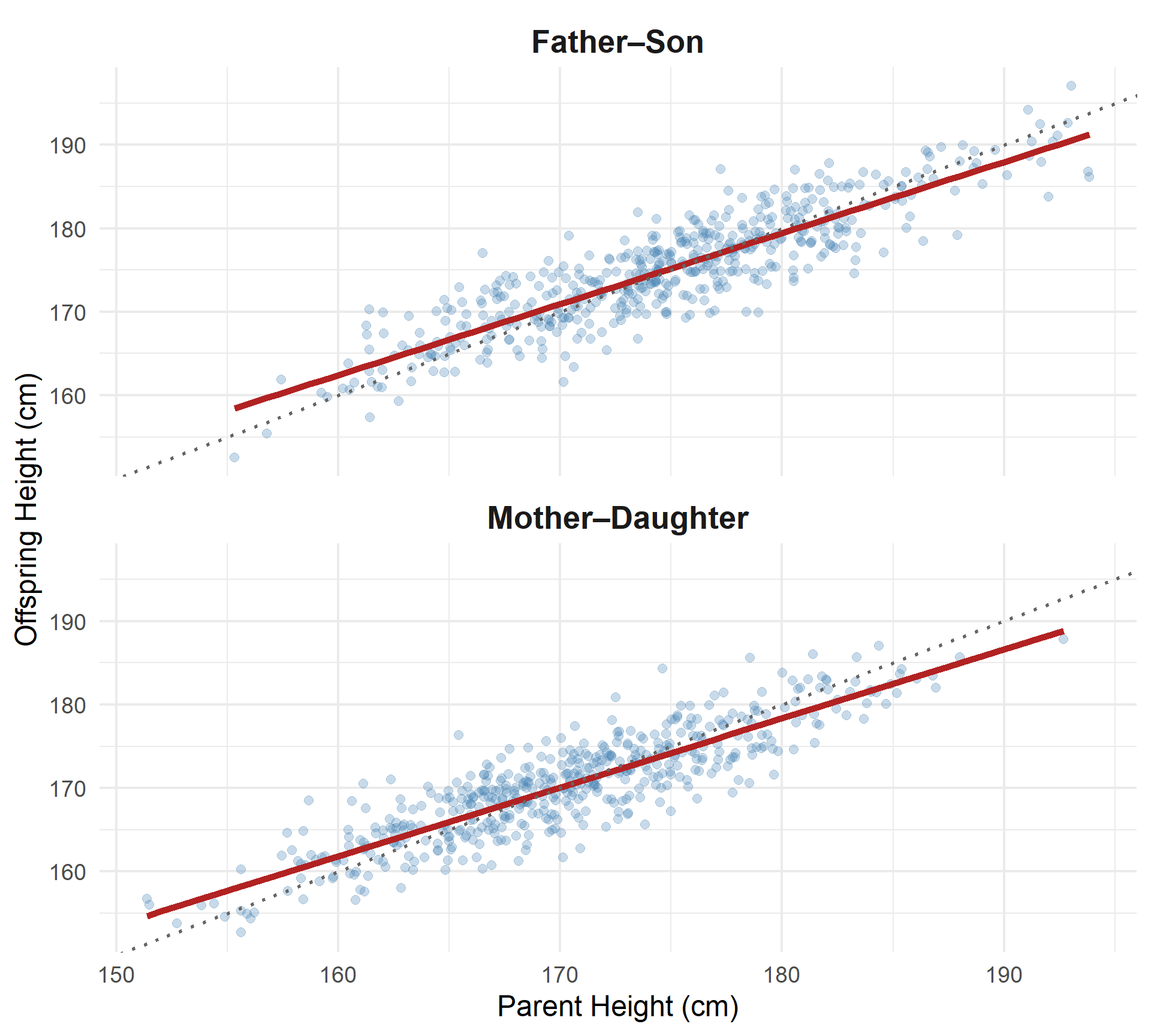
Parent–Offspring Regression for Height
- Each point = one parent–offspring pair
- Clear positive relationship
- Slope of regression line ≈ \(h^2\)
Interpretation:
The steeper the slope, the greater the additive genetic contribution to variation.
Selection Shifts the Population Mean
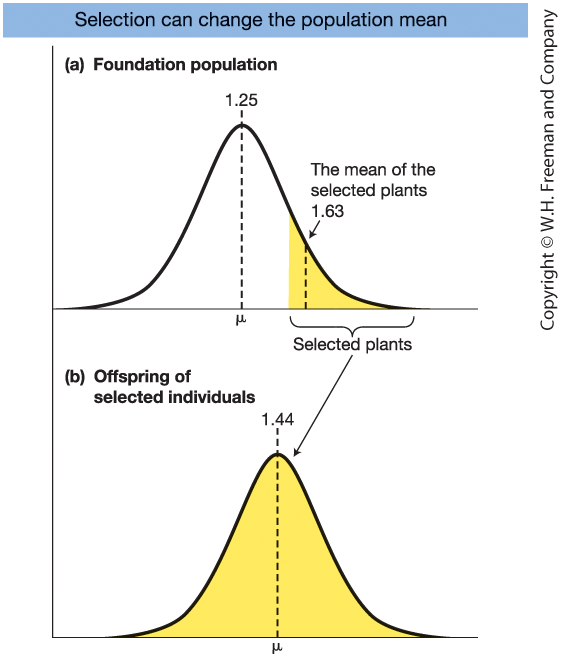
Figure 19-9.
One generation of artificial selection for provitamin A in maize.
Base population mean = 1.25 μg/g
Selected group mean = 1.63 μg/g
Offspring mean = 1.44 μg/g
Selection differential:
\[ S = 1.63 - 1.25 = 0.38 \]Selection response:
\[ R = 1.44 - 1.25 = 0.19 \]
Only the additive (heritable) component causes the shift in the next generation.
Long-Term Response to Artificial Selection
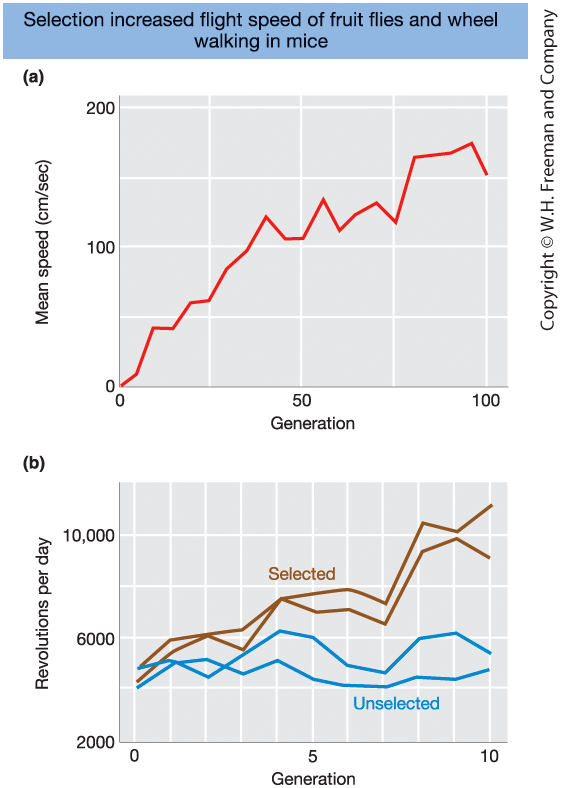
Figure 19-10.
Long-term selection experiments demonstrate sustained evolutionary change.
- Fruit flies
- Selected for increased flight speed
- Mean speed increased dramatically over 100 generations
- Mice
- Selected for voluntary wheel running
- Large increase over just 10 generations
- Control (unselected) lines changed very little
Key message:
Populations contain substantial additive genetic variation
and can respond to selection for many generations.
Why GWAS Works: Marker–QTL Linkage Disequilibrium
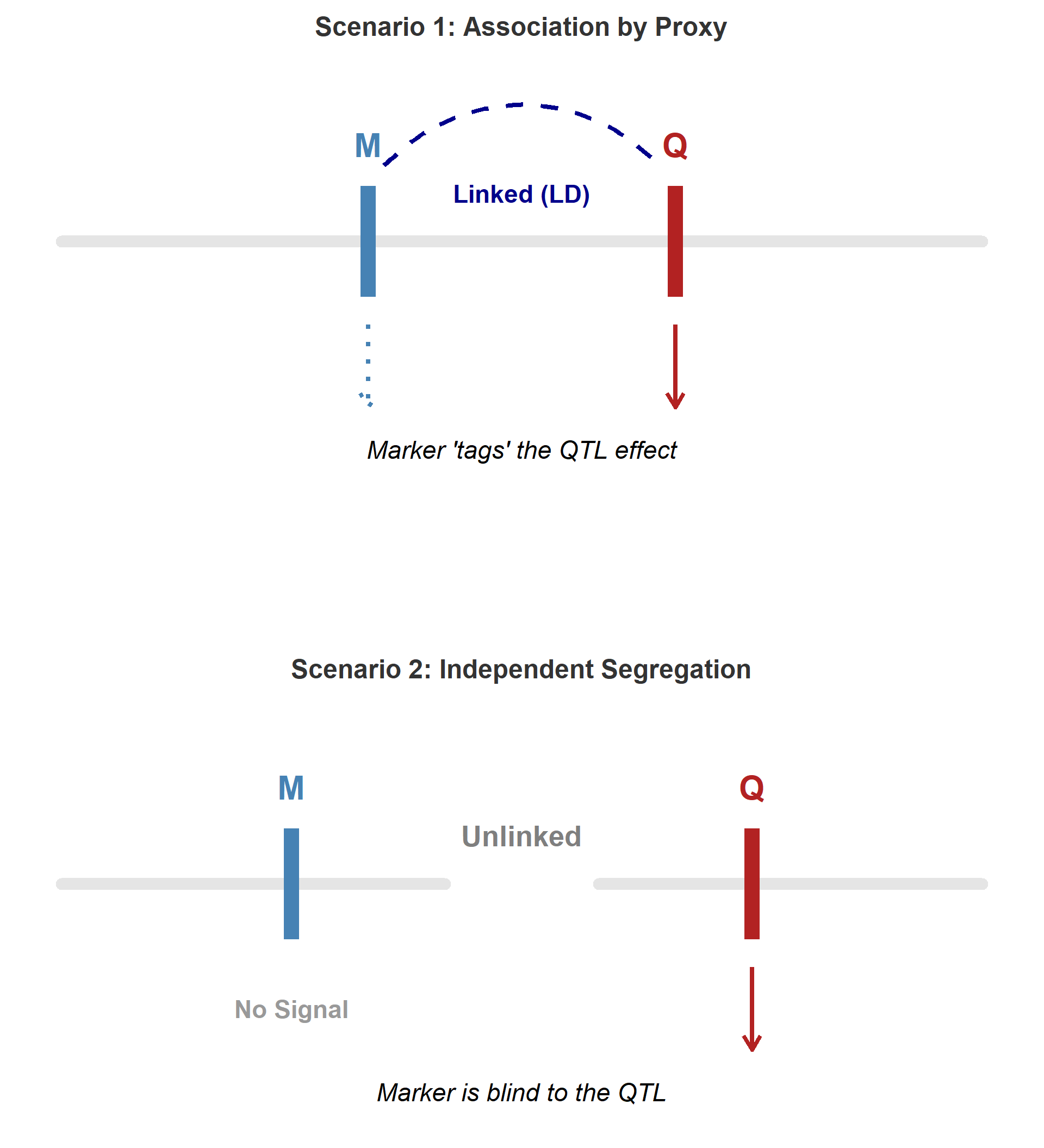
Concept
- A causal mutation (QTL) influences the trait
- A nearby SNP may be in linkage disequilibrium (LD) with the QTL
- The SNP is therefore correlated with the causal variant
Therefore, the SNP shows statistical association
even if it is not itself causal.
Key Point
GWAS detects markers that are in LD
with causal variants.
How GWAS Works: Single Marker Test
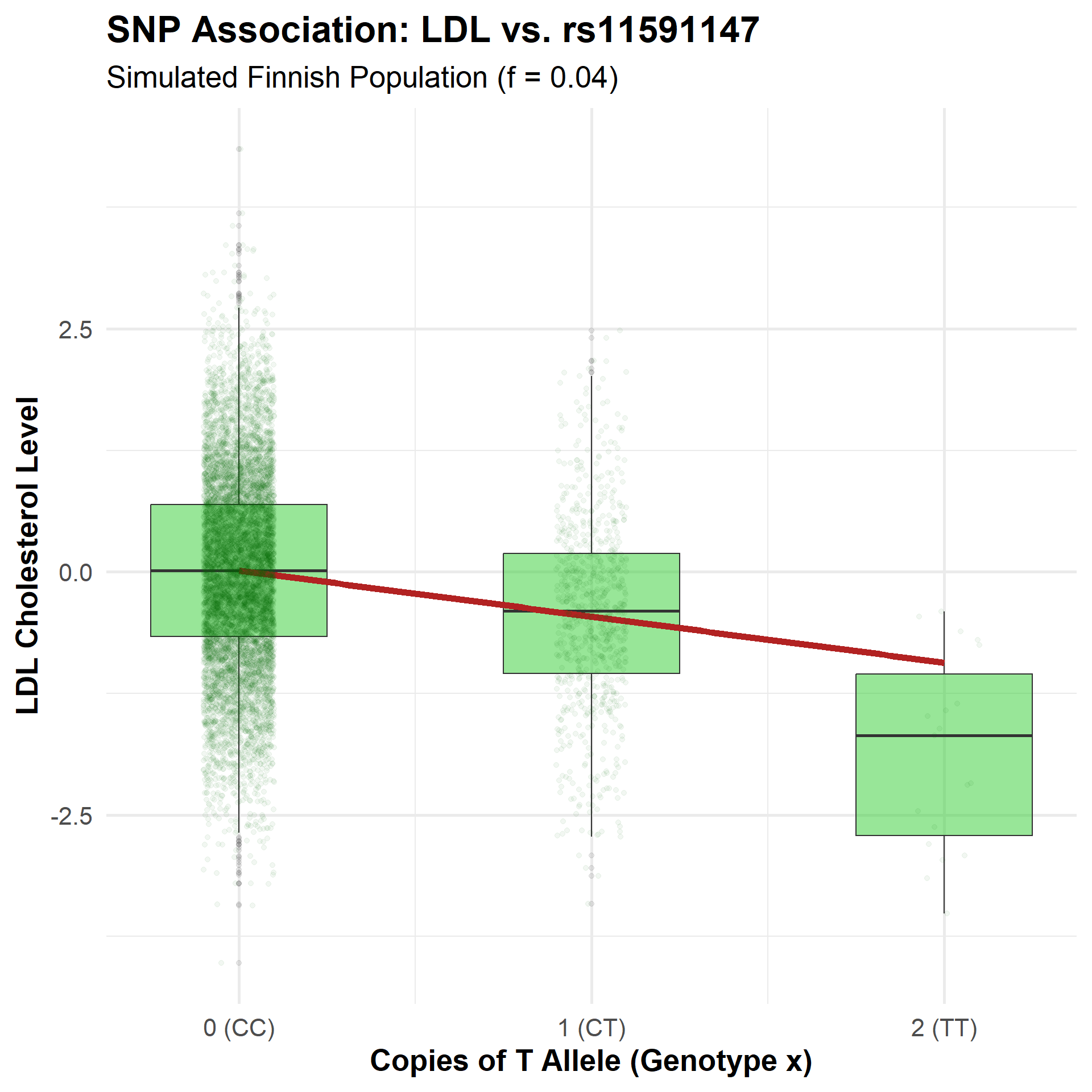
The Additive Model
We fit a linear regression: \[y = \mu + x\beta + \varepsilon\]
- \(y\): Phenotype (LDL)
- \(x\): Genotype (0, 1, or 2)
- \(\beta\): Effect size per allele
Interpretation
- \(\beta\) represents the average change in LDL for each additional T allele.
- The \(p\)-value from this model tests the null hypothesis: \(\beta = 0\).
- Here, each T allele significantly decreases LDL.
How GWAS Works: Many Marker Test
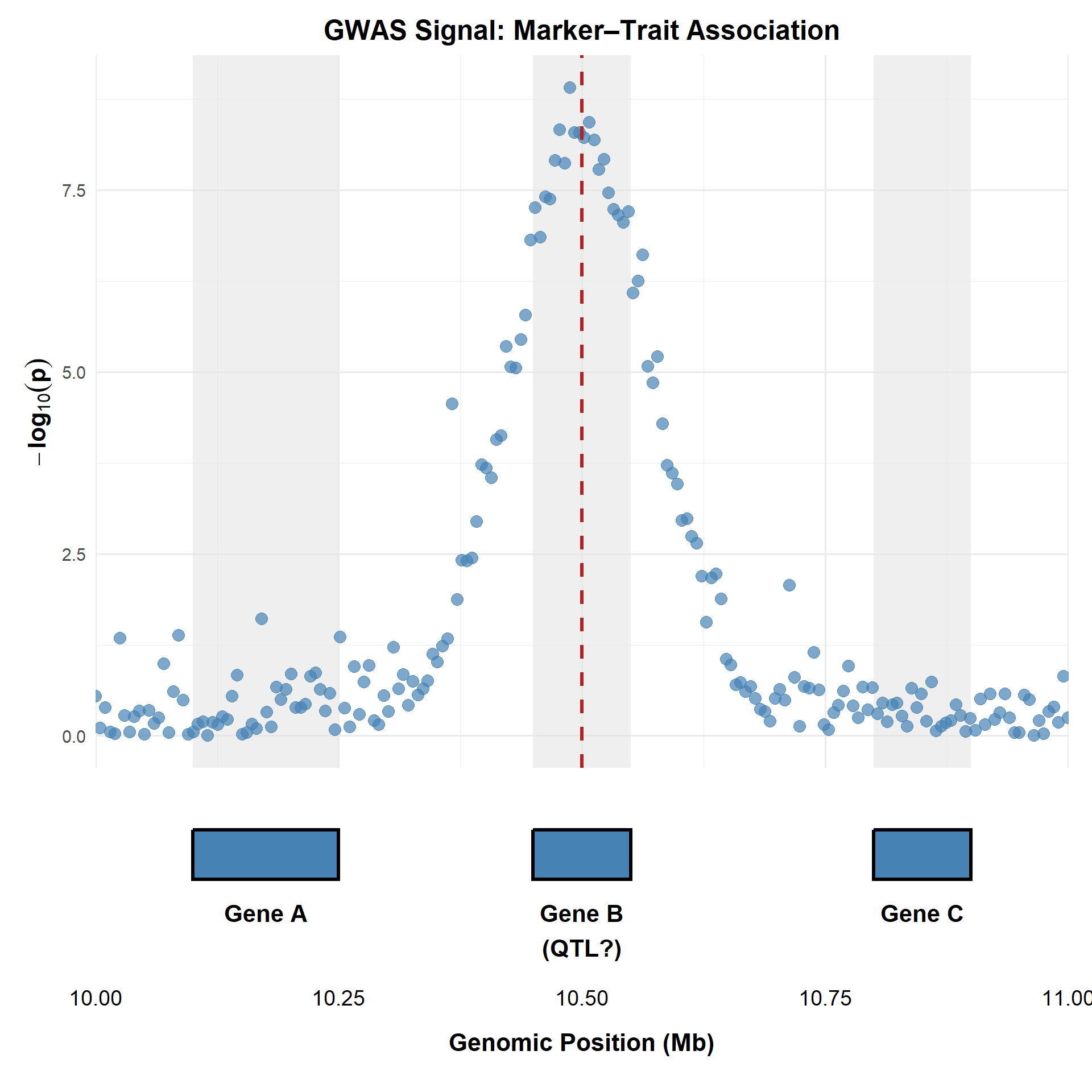
The Interpretation
- Association Peak: The highest points cluster over a specific genomic region.
- Candidate Gene (QTL): The vertical dashed line shows the “lead SNP” falls directly within Gene B.
- The “Neighborhood” Effect: Note how the shading spans the whole gene. Because of LD, many SNPs in the red-shaded region appear significant, but only one gene is likely the true functional driver.
Key point This visual demonstrates Fine-Mapping: the process of narrowing down a broad GWAS “skyscraper” to the specific gene region most likely to contain the causal variant.
How GWAS Works: From P-values to Manhattan Plot
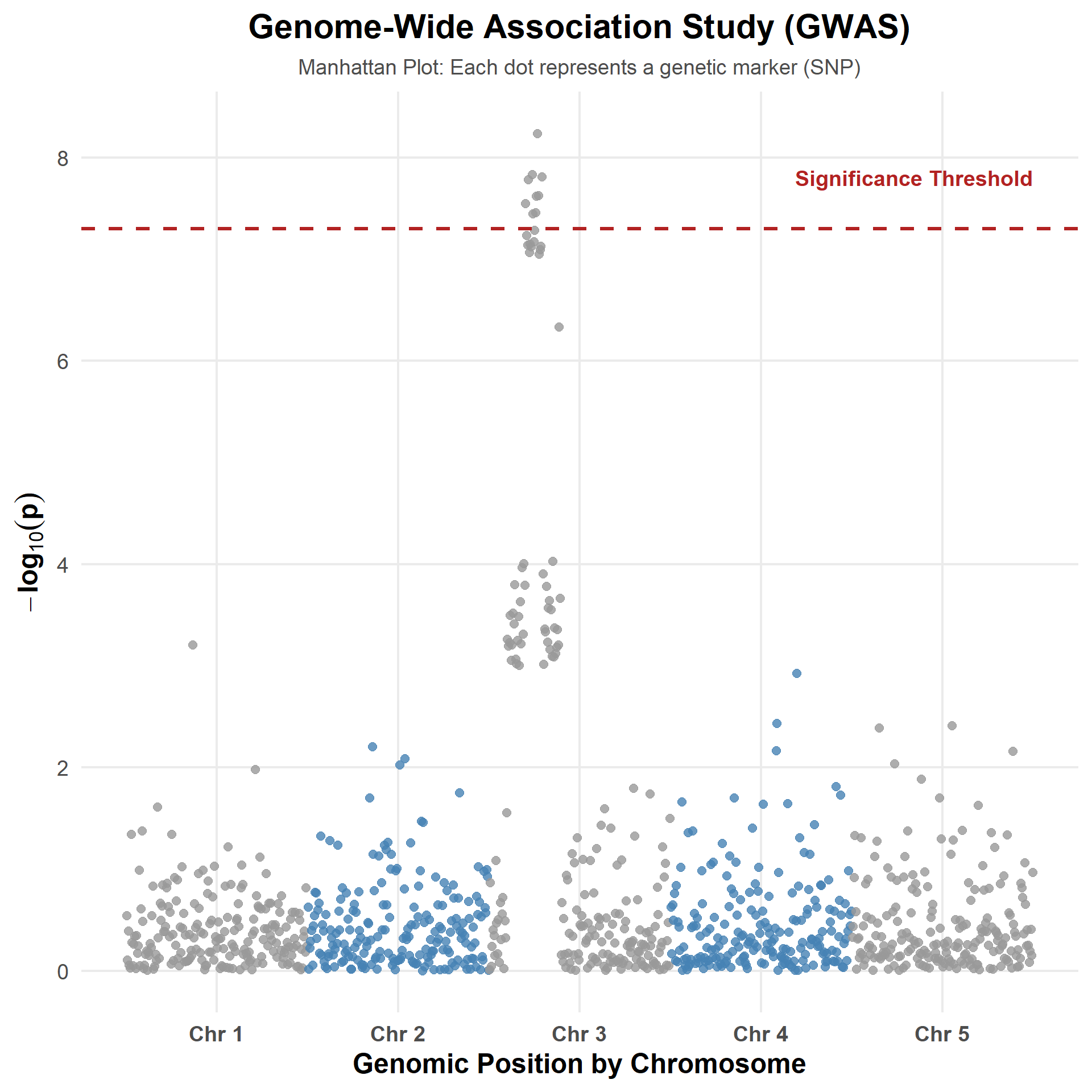
Each point = one SNP
- X-axis → genomic position
- Y-axis → \(-\log_{10}(p)\)
Higher points = stronger evidence
against the null hypothesis.
Dashed line
- Genome-wide significance threshold
\(\bigl(\text{e.g., } p < 5\times10^{-8}\bigr)\)
Peaks indicate genomic regions
associated with the trait.
GWAS Identifies a Gene for Body Size in Dogs
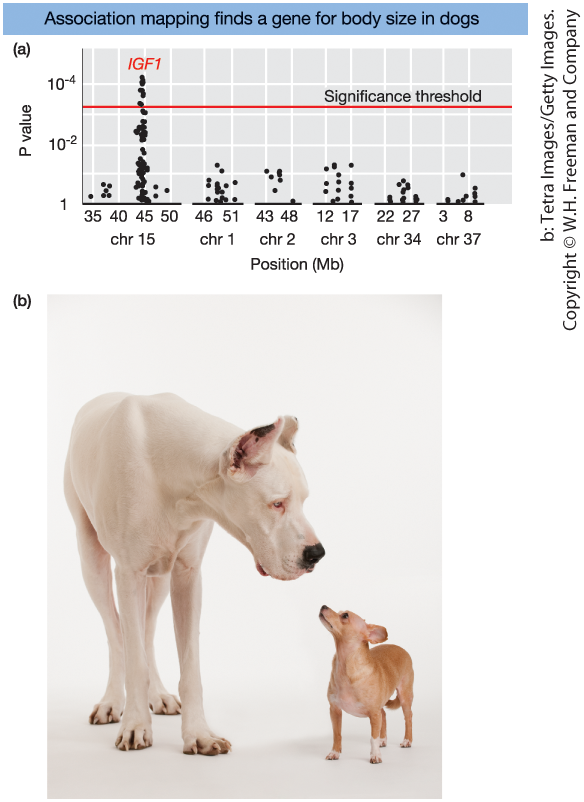
Figure 19-18.
- Genome-wide association study (GWAS) for body size in dogs
- Each dot = one SNP
- Y-axis = −log10(P value)
- Horizontal line = significance threshold
- Strong peak on chromosome 15
→ Region contains the IGF1 gene
- Small vs. large dog breeds
IGF1 is a major contributor to size differences.
GWAS Identifies Genes for Common Human Diseases
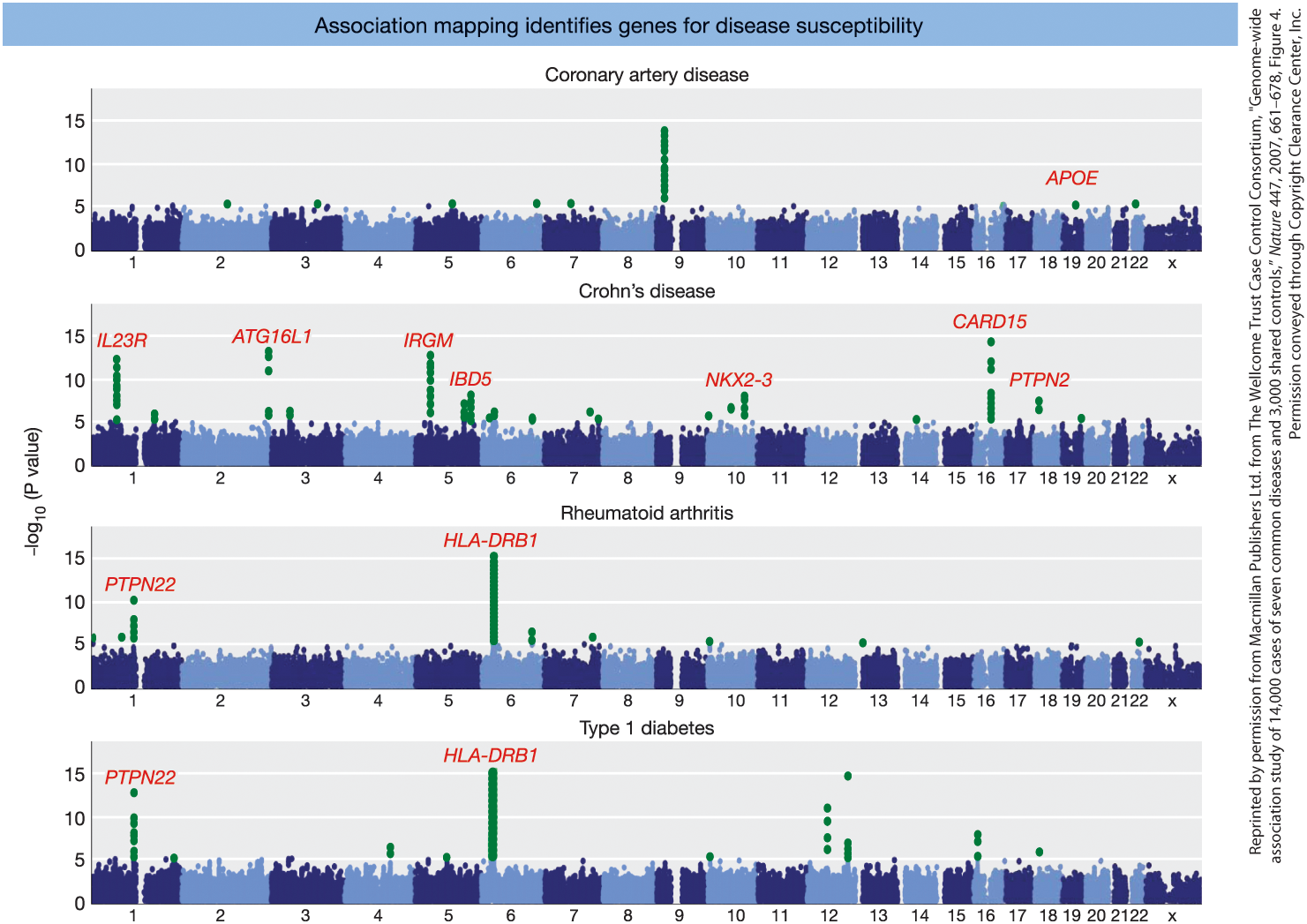
Figure 19-19.
Genome-wide association studies (GWAS)
for several common human diseases.
- X-axis: Chromosomes 1–22
- Y-axis: −log10(P value)
- Green dots = significant SNP associations
- Red labels = candidate genes identified
Examples:
- Coronary artery disease → APOE
- Crohn’s disease → IL23R, ATG16L1, IRGM
- Rheumatoid arthritis → PTPN22, HLA-DRB1
- Type 1 diabetes → PTPN22, HLA-DRB1